Autoencoders for RNA-seq data#
In this notebook we work with high-dimensional data, meaning we have many more features than samples. High dimensionality can be challenging because distances and similarities become less informative and models can overfit more easily (often summarized as the curse of dimensionality).
A common clinical data type with very high feature counts is sequencing data. In RNA-seq, each sample is represented by expression values for thousands of genes, so the input space is naturally large. Here we use an autoencoder to learn a compact latent representation of RNA-seq profiles that can be used for visualization and downstream analyses. The dataset we use for this is called CPTAC. CPTAC (the Clinical Proteomic Tumor Analysis Consortium) is an NCI-led effort that provides large, public multi-omics datasets from patient tumors, with matched clinical annotations.
Goal:
Load bulk RNA-seq data from CPTAC via the
cptacPython packageTrain an autoencoder to compress high-dimensional gene expression into a low-dimensional latent space
Visualize reconstruction loss and latent representations (PCA)
from google.colab import ai
response = ai.generate_text("Tell me what AI you are and can you help me with coding problems?")
print(response)
Hello!
I am a large language model, trained by Google.
**Can I help you with coding problems? Yes, absolutely!**
I can assist you with a wide range of coding-related tasks, including:
1. **Debugging Code:** Help you identify errors in your code and suggest fixes.
2. **Writing Code Snippets:** Generate code for specific functions, algorithms, or data structures.
3. **Explaining Concepts:** Clarify programming concepts, algorithms, design patterns, or specific language features.
4. **Optimizing Code:** Suggest ways to make your code more efficient, readable, or performant.
5. **Code Translation:** Help translate code from one programming language to another.
6. **Understanding Error Messages:** Break down complex error messages into understandable terms and suggest solutions.
7. **Best Practices:** Provide guidance on best coding practices for various languages and scenarios.
8. **Project Planning (Conceptual):** Discuss approaches to structuring your code or planning out a project.
**To get the best help, please provide:**
* **Your code:** The more complete and relevant, the better.
* **The programming language:** (e.g., Python, JavaScript, Java, C++, Go, etc.)
* **A clear description of the problem:** What are you trying to do? What's going wrong?
* **Any error messages:** Copy and paste them directly.
* **What you've tried so far:** This helps me understand your thought process and avoid suggesting solutions you've already dismissed.
**Important Note:** While I can generate and explain code, I cannot *execute* code myself, nor do I have real-time access to your local machine or external systems. My knowledge is based on the data I was trained on, so always test the code I provide in your own environment.
Feel free to share your coding problem!
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import torch
import torch.nn as nn
from torch.utils.data import DataLoader, TensorDataset
from sklearn.model_selection import train_test_split
from sklearn.preprocessing import StandardScaler
from sklearn.decomposition import PCA
import umap
1) Load CPTAC dataset (RNA-seq)#
We use the cptac package, which provides programmatic access to CPTAC cohorts as pandas DataFrames.
Notes:
On first use, the dataset may download and cache locally (can take a bit, depending on cohort).
CPTAC cohorts contain multiple omics layers; we focus on transcriptomics (RNA-seq).
!pip install cptac # should take around 2-3 min
import cptac
Collecting cptac
Downloading cptac-1.5.14-py3-none-any.whl.metadata (8.2 kB)
Requirement already satisfied: numpy>=1.16.3 in /usr/local/lib/python3.12/dist-packages (from cptac) (2.0.2)
Requirement already satisfied: pandas>=1.5.3 in /usr/local/lib/python3.12/dist-packages (from cptac) (2.2.2)
Requirement already satisfied: requests>=2.21.0 in /usr/local/lib/python3.12/dist-packages (from cptac) (2.32.4)
Requirement already satisfied: scipy>=1.10.0 in /usr/local/lib/python3.12/dist-packages (from cptac) (1.16.3)
Requirement already satisfied: openpyxl>=2.6.0 in /usr/local/lib/python3.12/dist-packages (from cptac) (3.1.5)
Requirement already satisfied: statsmodels>=0.10.0 in /usr/local/lib/python3.12/dist-packages (from cptac) (0.14.6)
Collecting pyranges>=0.0.111 (from cptac)
Downloading pyranges-0.1.4-py3-none-any.whl.metadata (3.7 kB)
Requirement already satisfied: tqdm>=4.65.0 in /usr/local/lib/python3.12/dist-packages (from cptac) (4.67.3)
Requirement already satisfied: et-xmlfile in /usr/local/lib/python3.12/dist-packages (from openpyxl>=2.6.0->cptac) (2.0.0)
Requirement already satisfied: python-dateutil>=2.8.2 in /usr/local/lib/python3.12/dist-packages (from pandas>=1.5.3->cptac) (2.9.0.post0)
Requirement already satisfied: pytz>=2020.1 in /usr/local/lib/python3.12/dist-packages (from pandas>=1.5.3->cptac) (2025.2)
Requirement already satisfied: tzdata>=2022.7 in /usr/local/lib/python3.12/dist-packages (from pandas>=1.5.3->cptac) (2025.3)
Collecting ncls>=0.0.63 (from pyranges>=0.0.111->cptac)
Downloading ncls-0.0.70-cp312-cp312-manylinux_2_17_x86_64.manylinux2014_x86_64.whl.metadata (4.9 kB)
Requirement already satisfied: tabulate in /usr/local/lib/python3.12/dist-packages (from pyranges>=0.0.111->cptac) (0.9.0)
Collecting sorted_nearest>=0.0.33 (from pyranges>=0.0.111->cptac)
Downloading sorted_nearest-0.0.41.tar.gz (1.6 MB)
━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 1.6/1.6 MB 29.3 MB/s eta 0:00:00
?25h Installing build dependencies ... ?25l?25hdone
Getting requirements to build wheel ... ?25l?25hdone
Preparing metadata (pyproject.toml) ... ?25l?25hdone
Requirement already satisfied: natsort in /usr/local/lib/python3.12/dist-packages (from pyranges>=0.0.111->cptac) (8.4.0)
Requirement already satisfied: charset_normalizer<4,>=2 in /usr/local/lib/python3.12/dist-packages (from requests>=2.21.0->cptac) (3.4.4)
Requirement already satisfied: idna<4,>=2.5 in /usr/local/lib/python3.12/dist-packages (from requests>=2.21.0->cptac) (3.11)
Requirement already satisfied: urllib3<3,>=1.21.1 in /usr/local/lib/python3.12/dist-packages (from requests>=2.21.0->cptac) (2.5.0)
Requirement already satisfied: certifi>=2017.4.17 in /usr/local/lib/python3.12/dist-packages (from requests>=2.21.0->cptac) (2026.2.25)
Requirement already satisfied: patsy>=0.5.6 in /usr/local/lib/python3.12/dist-packages (from statsmodels>=0.10.0->cptac) (1.0.2)
Requirement already satisfied: packaging>=21.3 in /usr/local/lib/python3.12/dist-packages (from statsmodels>=0.10.0->cptac) (26.0)
Requirement already satisfied: six>=1.5 in /usr/local/lib/python3.12/dist-packages (from python-dateutil>=2.8.2->pandas>=1.5.3->cptac) (1.17.0)
Downloading cptac-1.5.14-py3-none-any.whl (5.5 MB)
━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 5.5/5.5 MB 75.9 MB/s eta 0:00:00
?25hDownloading pyranges-0.1.4-py3-none-any.whl (1.5 MB)
━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 1.5/1.5 MB 68.7 MB/s eta 0:00:00
?25hDownloading ncls-0.0.70-cp312-cp312-manylinux_2_17_x86_64.manylinux2014_x86_64.whl (2.7 MB)
━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 2.7/2.7 MB 72.5 MB/s eta 0:00:00
?25hBuilding wheels for collected packages: sorted_nearest
Building wheel for sorted_nearest (pyproject.toml) ... ?25l?25hdone
Created wheel for sorted_nearest: filename=sorted_nearest-0.0.41-cp312-cp312-linux_x86_64.whl size=6699045 sha256=c4b54aa87e241b23849cf17756ea3e9dc5556c9be1a10d36259070d55a3fa44d
Stored in directory: /root/.cache/pip/wheels/7c/cb/a4/a6f831a60e479b9001be190e91000cc472addeab871957e5a9
Successfully built sorted_nearest
Installing collected packages: sorted_nearest, ncls, pyranges, cptac
Successfully installed cptac-1.5.14 ncls-0.0.70 pyranges-0.1.4 sorted_nearest-0.0.41
Downloading cptac_genes.csv: 100%|██████████| 462k/462k [00:25<00:00, 18.4kB/s]
Downloading brca_mapping.csv: 100%|██████████| 6.37k/6.37k [00:06<00:00, 958B/s]
Downloading index.tsv: 100%|██████████| 30.2k/30.2k [00:12<00:00, 2.46kB/s]
Now, lets see what data we have available in this package.
cptac.get_source_options()
| Cancer | ||
|---|---|---|
| Source | Datatype | |
| bcm | CNV | [brca, gbm, ov, ucec, pdac, coad, luad, ccrcc,... |
| circular_RNA | [hnscc, gbm, ucec, pdac, luad, ccrcc, lscc] | |
| miRNA | [hnscc, brca, gbm, ucec, pdac, coad, luad, ccr... | |
| phosphoproteomics | [hnscc, brca, gbm, ov, ucec, pdac, coad, luad,... | |
| proteomics | [hnscc, brca, gbm, ov, ucec, pdac, coad, luad,... | |
| transcriptomics | [hnscc, brca, gbm, ov, ucec, pdac, coad, luad,... | |
| broad | transcriptomics | [hnscc, brca, gbm, ov, ucec, pdac, coad, luad,... |
| harmonized | ancestry_prediction | [all_cancers] |
| somatic_mutation | [all_cancers] | |
| mssm | clinical | [all_cancers] |
| umich | acetylproteomics | [brca, gbm, ucec, luad, lscc] |
| phosphoproteomics | [hnscc, brca, gbm, ucec, pdac, coad, luad, ccr... | |
| proteomics | [hnscc, brca, gbm, ov, ucec, pdac, coad, luad,... | |
| washu | CNV | [hnscc, brca, gbm, ov, ucec, pdac, coad, luad,... |
| cibersort | [hnscc, brca, gbm, ov, ucec, pdac, coad, luad,... | |
| hla_typing | [all_cancers] | |
| mature_miRNA | [hnscc, gbm, ucec, pdac, luad, ccrcc, lscc] | |
| precursor_miRNA | [hnscc, gbm, ucec, pdac, luad, ccrcc, lscc] | |
| somatic_mutation | [hnscc, gbm, ov, ucec, pdac, coad, luad, lscc] | |
| total_miRNA | [hnscc, gbm, ucec, pdac, luad, ccrcc, lscc] | |
| transcriptomics | [hnscc, brca, gbm, ov, ucec, pdac, coad, luad,... | |
| tumor_purity | [all_cancers] | |
| xcell | [hnscc, brca, gbm, ov, ucec, pdac, coad, luad,... |
2) Get RNA-seq matrix#
CPTAC uses multiple data sources; transcriptomics is typically available as a dataframe with gene expressions as columns and patients as rows. We’ll:
extract transcriptomics counts for several cancer types
combine them into one big dataframe
drop samples with missing values
cohort_classes = [
("Brca", cptac.Brca),
("Ccrcc", cptac.Ccrcc),
("Colon", cptac.Coad),
("Gbm", cptac.Gbm),
("Luad", cptac.Luad),
("Ovarian", cptac.Ov),
("Pdac", cptac.Pdac),
("Ucec", cptac.Ucec),]
source = "bcm"
dfs = []
loaded = []
for name, cls in cohort_classes:
cohort = cls()
tx = cohort.get_transcriptomics(source)
tx = tx.copy()
tx["cancer_type"] = name
dfs.append(tx)
loaded.append((name, tx.shape))
print(f"Loaded {name}: {tx.shape}")
print("\nLoaded cohorts:", loaded)
Downloading BRCA-gene_RSEM_tumor_normal_UQ_log2(x+1)_BCM.txt.gz: 100%|██████████| 7.26M/7.26M [00:18<00:00, 400kB/s]
Downloading gencode.v34.basic.annotation-mapping.txt.gz: 100%|██████████| 1.75M/1.75M [00:14<00:00, 118kB/s]
Loaded Brca: (133, 60592)
Downloading CCRCC-gene_rsem_removed_circRNA_tumor_normal_UQ_log2(x+1)_BCM.txt.gz: 100%|██████████| 12.9M/12.9M [00:13<00:00, 983kB/s]
Downloading gencode.v34.basic.annotation-mapping.txt.gz: 100%|██████████| 1.75M/1.75M [00:27<00:00, 64.0kB/s]
Loaded Ccrcc: (185, 59287)
Downloading CRC-gene_RSEM_tumor_normal_UQ_log2(x+1)_BCM.txt.gz: 100%|██████████| 5.68M/5.68M [00:13<00:00, 407kB/s]
Downloading gencode.v34.basic.annotation-mapping.txt.gz: 100%|██████████| 1.75M/1.75M [00:14<00:00, 119kB/s]
Loaded Colon: (106, 60592)
Downloading GBM-gene_rsem_removed_circRNA_tumor_normal_UQ_log2(x+1)_BCM.txt.gz: 100%|██████████| 7.37M/7.37M [00:12<00:00, 608kB/s]
Downloading gencode.v34.basic.annotation-mapping.txt.gz: 100%|██████████| 1.75M/1.75M [00:10<00:00, 174kB/s]
Loaded Gbm: (100, 59287)
Downloading LUAD-gene_rsem_removed_circRNA_tumor_normal_UQ_log2(x+1)_BCM.txt.gz: 100%|██████████| 14.9M/14.9M [00:11<00:00, 1.32MB/s]
Downloading gencode.v34.basic.annotation-mapping.txt.gz: 100%|██████████| 1.75M/1.75M [00:12<00:00, 136kB/s]
Loaded Luad: (213, 59287)
Downloading OV-gene_RSEM_tumor_normal_UQ_log2(x+1)_BCM.txt.gz: 100%|██████████| 5.34M/5.34M [00:14<00:00, 376kB/s]
Downloading gencode.v34.basic.annotation-mapping.txt.gz: 100%|██████████| 1.75M/1.75M [00:11<00:00, 152kB/s]
Loaded Ovarian: (101, 60592)
Downloading PDAC-gene_rsem_removed_circRNA_tumor_normal_UQ_log2(x+1)_BCM.txt.gz: 100%|██████████| 14.7M/14.7M [00:20<00:00, 731kB/s]
Downloading gencode.v34.basic.annotation-mapping.txt.gz: 100%|██████████| 1.75M/1.75M [00:16<00:00, 108kB/s]
Loaded Pdac: (212, 59287)
Downloading UCEC-gene_rsem_removed_circRNA_tumor_normal_UQ_log2(x+1)_BCM.txt.gz: 100%|██████████| 8.50M/8.50M [00:16<00:00, 515kB/s]
Downloading gencode.v34.basic.annotation-mapping.txt.gz: 100%|██████████| 1.75M/1.75M [00:10<00:00, 166kB/s]
Loaded Ucec: (119, 59287)
Loaded cohorts: [('Brca', (133, 60592)), ('Ccrcc', (185, 59287)), ('Colon', (106, 60592)), ('Gbm', (100, 59287)), ('Luad', (213, 59287)), ('Ovarian', (101, 60592)), ('Pdac', (212, 59287)), ('Ucec', (119, 59287))]
dfs[0] # please have a look how many genes there are in this dataset
| Name | A1BG | A1BG-AS1 | A1CF | A2M | A2M-AS1 | A2ML1 | A2ML1-AS1 | A2ML1-AS2 | A2MP1 | A3GALT2 | ... | ZXDC | ZYG11A | ZYG11AP1 | ZYG11B | ZYX | ZYXP1 | ZZEF1 | hsa-mir-1253 | hsa-mir-423 | cancer_type |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Database_ID | ENSG00000121410.12 | ENSG00000268895.6 | ENSG00000148584.15 | ENSG00000175899.15 | ENSG00000245105.4 | ENSG00000166535.20 | ENSG00000256661.1 | ENSG00000256904.1 | ENSG00000256069.7 | ENSG00000184389.9 | ... | ENSG00000070476.15 | ENSG00000203995.10 | ENSG00000232242.2 | ENSG00000162378.13 | ENSG00000159840.16 | ENSG00000274572.1 | ENSG00000074755.15 | ENSG00000272920.1 | ENSG00000266919.3 | |
| Patient_ID | |||||||||||||||||||||
| 01BR001 | 3.51 | 8.04 | 0.00 | 12.93 | 5.01 | 4.88 | 0.0 | 0.0 | 3.13 | 0.00 | ... | 8.88 | 7.07 | 0.0 | 9.86 | 10.63 | 0.0 | 10.05 | 0.0 | 0.0 | Brca |
| 01BR008 | 2.84 | 8.24 | 2.50 | 13.02 | 5.67 | 10.10 | 0.0 | 0.0 | 0.00 | 0.00 | ... | 8.84 | 8.60 | 0.0 | 8.44 | 11.36 | 0.0 | 10.20 | 0.0 | 0.0 | Brca |
| 01BR009 | 4.85 | 8.11 | 0.00 | 13.68 | 5.87 | 9.50 | 0.0 | 0.0 | 3.11 | 0.00 | ... | 8.85 | 3.64 | 0.0 | 8.90 | 10.58 | 0.0 | 10.54 | 0.0 | 0.0 | Brca |
| 01BR010 | 4.24 | 7.96 | 0.00 | 12.64 | 4.82 | 5.03 | 0.0 | 0.0 | 1.33 | 0.00 | ... | 8.31 | 8.28 | 0.0 | 9.28 | 12.40 | 0.0 | 10.42 | 0.0 | 0.0 | Brca |
| 01BR015 | 3.89 | 7.56 | 1.58 | 13.55 | 5.32 | 3.31 | 0.0 | 0.0 | 2.99 | 0.00 | ... | 9.05 | 5.08 | 0.0 | 10.13 | 10.44 | 0.0 | 10.46 | 0.0 | 0.0 | Brca |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 21BR002 | 0.00 | 7.86 | 0.00 | 13.96 | 6.51 | 1.12 | 0.0 | 0.0 | 0.00 | 1.74 | ... | 9.79 | 5.47 | 0.0 | 8.97 | 11.72 | 0.0 | 10.18 | 0.0 | 0.0 | Brca |
| 21BR010 | 3.50 | 6.83 | 1.43 | 14.57 | 6.76 | 0.00 | 0.0 | 0.0 | 3.49 | 0.00 | ... | 9.14 | 4.63 | 0.0 | 11.22 | 8.95 | 0.0 | 10.19 | 0.0 | 0.0 | Brca |
| 22BR005 | 3.39 | 6.76 | 3.97 | 13.90 | 5.71 | 5.60 | 0.0 | 0.0 | 3.72 | 0.00 | ... | 9.07 | 7.72 | 0.0 | 9.64 | 10.20 | 0.0 | 10.19 | 0.0 | 0.0 | Brca |
| 22BR006 | 2.59 | 7.30 | 0.88 | 13.98 | 6.00 | 1.42 | 0.0 | 0.0 | 1.43 | 0.00 | ... | 8.81 | 6.19 | 0.0 | 9.52 | 11.82 | 0.0 | 10.45 | 0.0 | 0.0 | Brca |
| 604 | 0.00 | 8.06 | 0.00 | 12.73 | 7.04 | 10.55 | 0.0 | 0.0 | 2.67 | 1.48 | ... | 10.51 | 9.03 | 0.0 | 9.13 | 12.19 | 0.0 | 9.61 | 0.0 | 0.0 | Brca |
133 rows × 60592 columns
expr_dfs = [d.drop(columns=["cancer_type"]) for d in dfs]
labels = pd.concat([d["cancer_type"] for d in dfs], axis=0)
# Find common genes across cohorts
common_genes = set(expr_dfs[0].columns)
for d in expr_dfs[1:]:
common_genes &= set(d.columns)
common_genes = sorted(common_genes)
print("Common genes across loaded cohorts:", len(common_genes))
expr = pd.concat([d[common_genes] for d in expr_dfs], axis=0)
print("Combined expression shape:", expr.shape)
print("Labels shape:", labels.shape)
Common genes across loaded cohorts: 59286
Combined expression shape: (1169, 59286)
Labels shape: (1169,)
expr = expr.dropna(axis=0, how="any")
expr = expr.dropna(axis=1, how="any")
labels_all = labels.loc[expr.index]
3) Train/validation split + scaling#
Autoencoders are trained to reconstruct the input. We scale features so training is stable:
fit scaler on training set only
apply to validation set
y = labels_all.values
X = expr.values.astype(np.float32)
X_trainval, X_test, y_trainval, y_test = train_test_split(
X, y, test_size=0.2, random_state=42, stratify=y
)
X_train, X_val, y_train, y_val = train_test_split(
X_trainval, y_trainval, test_size=0.2, random_state=42, stratify=y_trainval
)
scaler = StandardScaler()
X_train_s = scaler.fit_transform(X_train).astype(np.float32)
X_val_s = scaler.transform(X_val).astype(np.float32)
X_test_s = scaler.transform(X_test).astype(np.float32)
train_ds = TensorDataset(torch.tensor(X_train_s), torch.tensor(X_train_s))
val_ds = TensorDataset(torch.tensor(X_val_s), torch.tensor(X_val_s))
test_ds = TensorDataset(torch.tensor(X_test_s), torch.tensor(X_test_s))
train_loader = DataLoader(train_ds, batch_size=32, shuffle=True)
val_loader = DataLoader(val_ds, batch_size=64, shuffle=False)
test_loader = DataLoader(test_ds, batch_size=64, shuffle=False)
input_dim = X_train_s.shape[1]
print("Input dim:", input_dim)
print("Train/Val/Test:", X_train_s.shape[0], X_val_s.shape[0], X_test_s.shape[0])
Input dim: 59286
Train/Val/Test: 748 187 234
4) Define an autoencoder#
An autoencoder is a neural network trained to reconstruct its input. It consists of an encoder that compresses the data into a low-dimensional latent embedding, and a decoder that reconstructs the original data from that embedding.
We use autoencoders to learn compact representations of high-dimensional data (e.g., gene expression) in an unsupervised way. The latent embeddings can then be used for visualization (PCA/UMAP), clustering, denoising, or as features for downstream prediction models.

class Autoencoder(nn.Module):
def __init__(self, input_dim, hidden_dim1=4096, hidden_dim2=1024, latent_dim=512):
super().__init__()
self.encoder = nn.Sequential(
nn.Linear(input_dim, hidden_dim1),
nn.ReLU(),
nn.Linear(hidden_dim1, hidden_dim2),
nn.ReLU(),
nn.Linear(hidden_dim2, latent_dim),
)
self.decoder = nn.Sequential(
nn.Linear(latent_dim, hidden_dim2),
nn.ReLU(),
nn.Linear(hidden_dim2, hidden_dim1),
nn.ReLU(),
nn.Linear(hidden_dim1, input_dim),
)
def forward(self, x):
z = self.encoder(x)
x_hat = self.decoder(z)
return x_hat, z
In this case, our loss is the Mean Squared Error (MSE). MSE is used when the model is predicting continuous-valued outputs, because it penalizes how far each predicted value is from the target value. In an autoencoder, the target is the original input vector itself (e.g., gene expression levels), so the decoder is doing regression of continuous features rather than predicting discrete class labels.
device = torch.device("cuda")
print("Device:", device)
model = Autoencoder(input_dim=input_dim).to(device)
criterion = nn.MSELoss()
optimizer = torch.optim.Adam(model.parameters(), lr=1e-3)
print(model)
Device: cuda
Autoencoder(
(encoder): Sequential(
(0): Linear(in_features=59286, out_features=4096, bias=True)
(1): ReLU()
(2): Linear(in_features=4096, out_features=1024, bias=True)
(3): ReLU()
(4): Linear(in_features=1024, out_features=512, bias=True)
)
(decoder): Sequential(
(0): Linear(in_features=512, out_features=1024, bias=True)
(1): ReLU()
(2): Linear(in_features=1024, out_features=4096, bias=True)
(3): ReLU()
(4): Linear(in_features=4096, out_features=59286, bias=True)
)
)
5) Training loop#
def run_epoch_ae(model, loader, criterion, optimizer=None, device="cpu"):
is_train = optimizer is not None
model.train() if is_train else model.eval()
total_loss = 0.0
n = 0
for xb, yb in loader:
xb = xb.to(device)
yb = yb.to(device)
if is_train:
optimizer.zero_grad()
with torch.set_grad_enabled(is_train):
x_hat, _ = model(xb)
loss = criterion(x_hat, yb)
if is_train:
loss.backward()
optimizer.step()
total_loss += loss.item() * xb.size(0)
n += xb.size(0)
return total_loss / n
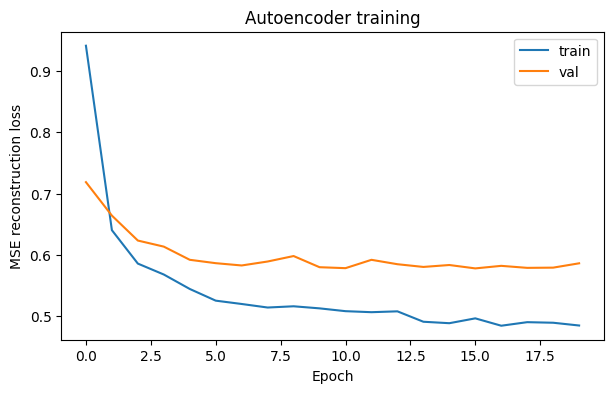
num_epochs = 20
train_losses = []
val_losses = []
for epoch in range(1, num_epochs + 1):
tr_loss = run_epoch_ae(model, train_loader, criterion, optimizer=optimizer, device=device)
va_loss = run_epoch_ae(model, val_loader, criterion, optimizer=None, device=device)
train_losses.append(tr_loss)
val_losses.append(va_loss)
if epoch == 1 or epoch % 10 == 0 or epoch == num_epochs:
print(f"Epoch {epoch:03d}/{num_epochs} | train_loss={tr_loss:.4f} | val_loss={va_loss:.4f}")
Epoch 001/20 | train_loss=0.9412 | val_loss=0.7188
Epoch 010/20 | train_loss=0.5130 | val_loss=0.5801
Epoch 020/20 | train_loss=0.4851 | val_loss=0.5865
6) Plot reconstruction loss#
plt.figure(figsize=(7,4))
plt.plot(train_losses, label="train")
plt.plot(val_losses, label="val")
plt.xlabel("Epoch")
plt.ylabel("MSE reconstruction loss")
plt.title("Autoencoder training")
plt.legend()
plt.show()
7) Extract latent space + PCA visualization#
We encode samples into a latent vector z (e.g. 128 dims),
then project to 2D with PCA for visualization.
@torch.no_grad()
def get_latent(model, X_scaled, device="cpu", batch_size=512):
model.eval()
zs = []
loader = DataLoader(torch.tensor(X_scaled, dtype=torch.float32),
batch_size=batch_size, shuffle=False)
for xb in loader:
xb = xb.to(device)
_, z = model(xb)
zs.append(z.cpu())
return torch.cat(zs, dim=0).numpy()
Z_test = get_latent(model, X_test_s, device=device)
print("Z_train shape:", Z_test.shape)
Z_train shape: (234, 512)
pca = PCA(n_components=2, random_state=42)
Z2 = pca.fit_transform(Z_test)
plt.figure(figsize=(6,5))
for ct in np.unique(y_test):
m = (y_test == ct)
plt.scatter(Z2[m, 0], Z2[m, 1], s=15, alpha=0.7, label=ct)
plt.xlabel("PC1")
plt.ylabel("PC2")
plt.title("PCA - Clusters of cancer types")
plt.legend(bbox_to_anchor=(1.02, 1), loc="upper left", markerscale=1.5)
plt.tight_layout()
plt.show()
print("Explained variance (PC1+PC2):", pca.explained_variance_ratio_.sum().round(3))
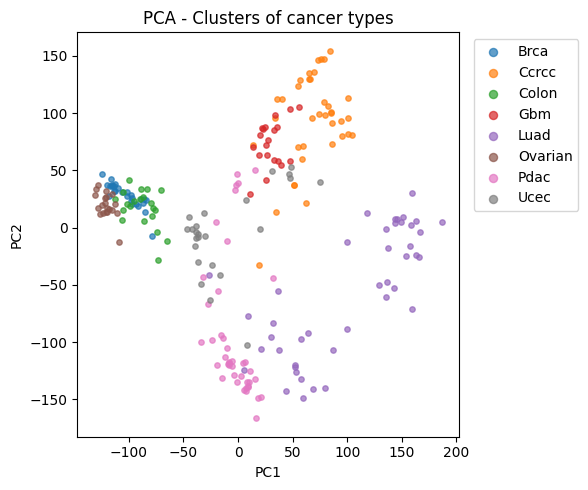
Explained variance (PC1+PC2): 0.326
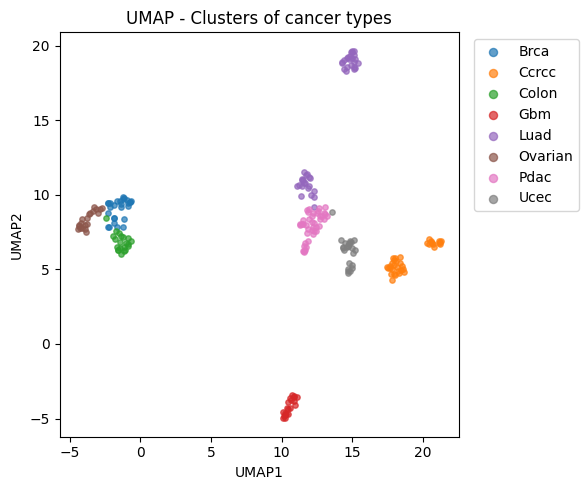
reducer = umap.UMAP(
n_neighbors=15,
min_dist=0.3,
metric="euclidean",
random_state=42
)
Z2 = reducer.fit_transform(Z_test)
plt.figure(figsize=(6,5))
for ct in np.unique(y_test):
m = (y_test == ct)
plt.scatter(Z2[m, 0], Z2[m, 1], s=15, alpha=0.7, label=ct)
plt.xlabel("UMAP1")
plt.ylabel("UMAP2")
plt.title("UMAP - Clusters of cancer types")
plt.legend(bbox_to_anchor=(1.02, 1), loc="upper left", markerscale=1.5)
plt.tight_layout()
plt.show()
/usr/local/lib/python3.12/dist-packages/umap/umap_.py:1952: UserWarning: n_jobs value 1 overridden to 1 by setting random_state. Use no seed for parallelism.
warn(